Doença de graves
Sintomas
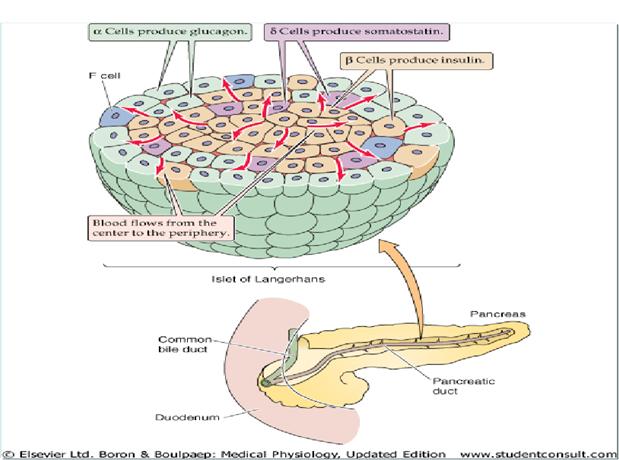
A doença de Graves é caracterizada clinicamente por hipertireoidismo e bócio, os pacientes geralmente apresentam sinais ou sintomas de hipertireoidismo (palpitações, taquicardia, arritmias, intolerância ao calor, aumento do apetite com perda de peso, diarreia, fraqueza e miopatia proximal, nervosismo e tremor).
Obs.: ocorre um aumento da vascularização da tireoide acompanhado de hiperplasia e hipertrofia do tecido endócrino.


Imagem disponível em:< http://www.medicodeolhos.com.br/2011/04/hipertireoidismo-proptose-exoftalmia.html > Acesso em 18/02/2013
Patogênese

Obs.: O IFN-α administrado como um agente antiviral, por exemplo, a pacientes que não conseguem curar a hepatite C, provavelmente tem efeito potente na ativação bystander uma vez que mimetiza o efeito de células dendríticas ativadas.
Obs.: O Campath-1H, um anticorpo monoclonal dirigido para um determinante de células T, foi administrado como um medicamento experimental a pacientes com a doença autoimune esclerose múltipla. Acredita-se que o medicamento gerou potente depleção de células T reguladoras de modo a permitir o aparecimento da autorreatividade.
A doença de Graves é caracterizada por autoanticorpos que estimulam a função glandular resultando em hipertireoidismo. Esses anticorpos são: um fator diferente do TSH e promovem a síntese e liberação dos hormônios tireoidianos, imunoglobulinas da classe IgG (esse fato foi descoberto devido à observação de que lactentes nascidos de mães com altos níveis séricos do anticorpo exibiam um hipertireoidismo transitório como resultado da transferência desses anticorpos pela placenta). Esses autoanticorpos competem com o TSH pela ligação ao receptor de TSH, por isso são atualmente denominados autoanticorpos do receptor de TSH (TRAb).
Como o diabetes tipo 1, a doença de Graves e o hipotireoidismo autoimune estão associados a polimorfismos no gene CTLA-4, é provável que exista entre essas doenças uma base subjacente semelhante; a partir dessa suposição pode-se inferir que a doença de graves possui uma tendência de gerar a autoimunidade das células T contra a tireoide a partir de moléculas de HLA-DR e HLA-DQ específicas e que a doença é facilitada pela função reguladora inadequada na periferia.

Modelo de doença para o desenvolvimento de tireoidite. A apresentação de autoanticorpos por APCs profissionais inicia o processo, que possui células TH1 e TH2. Quando o processo é estabelecido o próprio tireócito suprarregula moléculas imunes como HLA de classe II e se torna capaz de perpetuar a inflamação apresentando autoantígenos da própria tireoide às células T infiltradas. A lesão do tireócito provavelmente é o resultado dos efeitos combinados de linfócitos TCD4 e TCD8 e autoanticorpos. O processo pode ser acelerado por agentes externos como IFN-α. As células Treg podem ser defeituosas, uma vez que a doença está ligada a polimorfismos do gene CTLA-4, um regulador negativo das células T. Fonte: Imunologia: básica e clínica/ Mark Peakman, Diego Vergani; [tradução Elisianne Nopper… et al.].-Rio de Janeiro: Elsevier 2011.
Diagnóstico
Os achados laboratoriais consistem em elevação dos hormônios tireoidianos – tiroxina (T4) e tri-iodotironina (T3) – com supressão dos níveis do hormônio estimulante da tireoide (TSH).
O autoanticorpos, envolvido na doença, pode ser detectado através de duas técnicas: um ensaio de radioligando, no qual ele compete com o TSH, marcado com isótopo radioativo pela ligação com o receptor; ou em um ensaio biológico que avalia os efeitos estimulantes dos anticorpos em células de tireoide em cultura.
O diagnóstico é ratificado por uma cintilografia que demonstra captação difusa.
Obs.: A cintilografia da tireoide é um procedimento de Medicina Nuclear que consiste na obtenção de imagens da glândula tireóidea após a administração de radiofármacos (pertecnetato-Tc99m, iodo-131 ou iodo-123).Geralmente é realizada em conjunto com o cálculo da captação de I-131 ou I-123, que consiste na medida da porcentagem da dose de iodo radioativo administrada que se acumula na tireoide, em tempos selecionados após a ingestão do radioiodo.
Tratamento
O hipertireoidismo geralmente é controlado farmacologicamente, e a resposta à terapia está associada a um declínio dos autoanticorpos contra o receptor de TSH. Os agentes usados, carbimazol e propiltiouracil, inibem a síntese dos hormônios tireoidianos. Se ocorrer recorrência dos autoanticorpos pode ser necessária à realização de cirurgia ou ablação da tireoide com iodo radioativo.
As DAT (Drogas Antitireoidianas) continuam como tratamento de primeira escolha em pacientes com doença leve, bócios pequenos, crianças e adolescentes, e em situações especiais como na gravidez. Por outro lado, o 131I tem sido cada vez mais utilizado, porque é considerado um tratamento seguro, definitivo e de fácil aplicação. O risco de exacerbação do hipertireoidismo após administração do 131I, os fatores prognósticos de falência e o cálculo da dose administrada têm sido alguns dos aspectos discutidos na literatura recentemente.
Bibliografia
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-27302001000600014
http://www.cermen.com.br/exame-cintilografia-tireoide.php
http://www.medicodeolhos.com.br/2011/04/hipertireoidismo-proptose-exoftalmia.html